Tutorial
Here we quickly shows the use of Hoea without installation
1, ho = what?
Gene ontology and KEGG orthology are hierarchical structured, so the 'ho' here represnts the hierarchical ontology. One ho have a definition, and have many relationships with other ho.
2, Get Hoea tarbar source from Download. Decompress it. Change to the new created Hoea-[version] directory. ( $ as the shell command prompt, same as below).
$ wget http://sourceforge.net/projects/hoea/files/0.1.2/Hoea-[version].tar.gz/download $ tar zxvf Hoea-[version].tar.gz $ cd Hoea-[version]
3, There is some executable files of this module, and we have also prepared some sample data for illustration. So change to the src/Hoea/sample directory.
$ cd src/Hoea/sample/
4, For an Hoea analysis, you actually need 2 files. The sample data were taken from BMC Genomics. 2009 Nov 11;10(1):517, which contains microarray data of model legume Medicago truncatula under salt stress. Input for Hoea are the two files in sample directory, mt_probe_go.txt and probe_6h_up.txt.
mt_probe_go.txt file contains Gene ontology annotation of probe sets of Medicago truncatula microarry, part of the files looks like:
Mtr.32952.1.S1_s_at GO:0016301 Mtr.32952.1.S1_s_at GO:0006014 Mtr.32952.1.S1_s_at GO:0009507 Mtr.51583.1.S1_at GO:0008270 Mtr.51583.1.S1_at GO:0003676 Mtr.51583.1.S1_at GO:0005622 Mtr.6693.1.S1_s_at GO:0009658 Mtr.6693.1.S1_s_at GO:0005198 Mtr.6693.1.S1_s_at GO:0016226 Mtr.6693.1.S1_s_at GO:0009507this is a two column file, the first column is the probe set, the second is the GO terms which annotated to the correspond probe set.
the probe_6h_up.txt file contails probe sets selected from up-regulated probe sets at time point 6 hour, it is looks like:
Mtr.10036.1.S1_at Mtr.10151.1.S1_s_at Mtr.10029.1.S1_s_at Mtr.10159.1.S1_at Mtr.10063.1.S1_at Mtr.10121.1.S1_at Mtr.10112.1.S1_at Mtr.10141.1.S1_at Mtr.10122.1.S1_at Mtr.10045.1.S1_atthis file contains one probe set per line.
5, after we prepare those 3 things we need, we could start the Hoea run, notice that we located at the sample directory right now. just run the following command:
$ python ../Hoea.py -a mt_probe_go.txt -i probe_6h_up.txtsome information will print to the screen:
read in ho relationship file... done read in ho definition file... done read in background ho annotations... done generate ho term to gene id mapping,this may take a little while... done start analysis... input 520 items for analysis, 366 items with annotations 18248 items have annotations in background All Done!also, some additional file were generated in current directory. the name of these files all start with the input analysis file probe_6h_up.txt, files contains now include:
mt_probe_go.txt probe_6h_up.txt_GO_1.png probe_6h_up.txt_GO_3.dot probe_6h_up.txt_Rcom.Rout probe_6h_up.txt probe_6h_up.txt_GO_2.dot probe_6h_up.txt_GO_3.pdf probe_6h_up.txt_Rinput.txt probe_6h_up.txt_GO_1.dot probe_6h_up.txt_GO_2.pdf probe_6h_up.txt_GO_3.png probe_6h_up.txt_Routput.txt probe_6h_up.txt_GO_1.pdf probe_6h_up.txt_GO_2.png probe_6h_up.txt_Rcom.R
5, explanation of the command. in last step, we run the command python ../Hoea.py -a mt_probe_go.txt -i probe_6h_up.txt -t 50900 to finish the analysis. when -a option specifies the annotation file, -i option specifies the input file for analysis, the -t option specifies the total number. the script Hoea.py support many other options like:
$ python ../Hoea.py -h
Usage: Hoea.py -i input file -r ho.relation -d ho.definition -a annotation_file -s output_prefix -p pvalue_cutoff -n number_cutoff
Options:
--version show program's version number and exit
-h, --help show this help message and exit
-i INPUT, --input=INPUT
input file for analysis
-r HOREL, --ho-relation=HOREL
ho relation file
-d HODEF, --ho-definition=HODEF
ho definition file
-a ANNOTATION, --annotation=ANNOTATION
annotation file
-p PVALUE, --pvalue=PVALUE
FDR adjust P-value cutoff of ho terms [default: 0.05]
-n NUMBER, --number=NUMBER
number cutoff of an ho term with iterm assigned
[default: 5]
-s PREFIX, --prefix=PREFIX
prefix of output file [default: input filename]
6, explanation of the output. the final analysis result was stored in the file probe_6h_up.txt_Routput.txt, this is a tab-delimited file. One line of this file looks like:
GO:0022622 root system development 3 520 0.0032416502947 165 Mtr.12392.1.S1_at//Mtr.15877.1.S1_at//Mtr..12155.1.S1_at 0.238860994920602 1every row of this file starts with a ho term, like GO:0022622, the following fields are the description of this term, probe sets assigned to this terms in input file, total number of probe sets in input file, annotation rate of this term (165/50900), probe sets assigned to this terms in the annotation file, '//' seperated probe sets belong to this term, p-value, FDR-adjust p-value (Q-value), respectively.
also, some graphs were generated during the analysis, the probe_6h_up.txt_GO_1.png illustrated the enrichment situation of probe sets belong to the biological_process term: GO:0008150
part of this graph:

the fill color of each box means the statistical significant level, explained below:
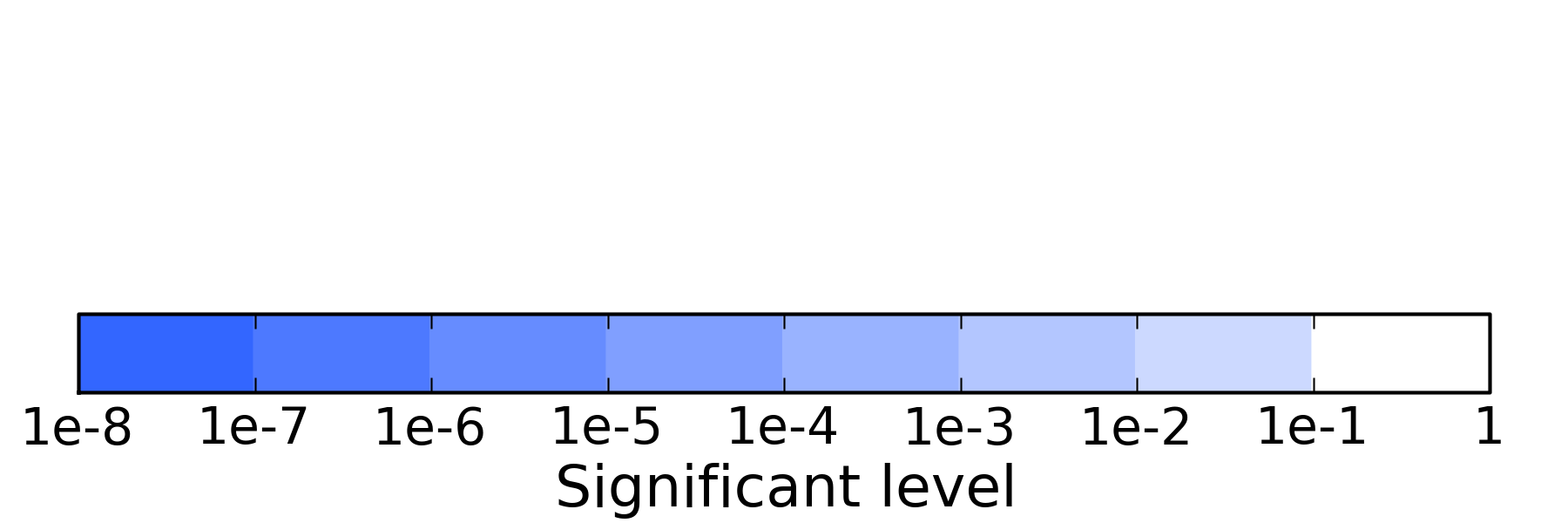
the relationship between each term were connected by lines with different color, blue means 'is_a', purple means 'regulates',
the color used was determinted by GO.relations from the Gene Ontology website.
other files like *.R file are the R scripts used for statistical calculation, the *.dot file are used by Graphviz for drawing those graphs.
7, update the ho relationship files and definition files
change to the data directory of Hoea module:
$ cd ../data/the files contains in the directory are:
go.def go.rel ko.def ko.relthe .def file means ho definition file and .rel file means ho relationship file.
as the Gene ontology and KEGG organization update their files frequently, Hoea contains two scripts GOIterator.py and KOIterator.py to help generating newest definition and relationship file.
$ python GOIterator.py -h
Usage: GOIterator.py -g gene_ontology_ext.obo -r go.relation -d go.definition
Options:
--version show program's version number and exit
-h, --help show this help message and exit
-g GOFILE, --go-file=GOFILE
specify the GO file
-r OUTREL, --output-relation=OUTREL
output GO relation file
-d OUTDEF, --output-difinition=OUTDEF
output GO definition file
the newest gene_ontology_ext.obo file is always available at: ftp://ftp.geneontology.org/go/ontology/obo_format_1_2/gene_ontology_ext.obo
$ python KOIterator.py -h
Usage: KOIterator.py -k ko -r ko.relation -d ko.definition
Options:
--version show program's version number and exit
-h, --help show this help message and exit
-k KOFILE, --ko-file=KOFILE
specify the KO file
-r OUTREL, --output-relation=OUTREL
output KO relation file
-d OUTDEF, --output-difinition=OUTDEF
output KO definition file
the newest ko file is always available at: ftp://ftp.genome.jp/pub/kegg/genes/ko
Next,we will show a KEGG orthology enrichment analysis example
this sample data were taken from a de novo transcriptome sequencing project of Medicago falcata (unpublished). the first 10 lines of annotation files looks like:
1_scaffold_10016-1 K12867 1_scaffold_19693-0 K08803 1_scaffold_4523-0 K02149 1_scaffold_25008-0 K10257 1_scaffold_22510-0 K04077 1_scaffold_17237-0 K03063 1_scaffold_5829-1 K11975 1_scaffold_13429-0 K10712 1_scaffold_4963-0 K01714 1_scaffold_20563-0 K13468this file is also a two column file, the first column indicate the transcripts id (from de novo assembly), the second column is the KO annotation terms. let's select some transcripts for enrichment analysis using Hoea.
2_scaffold_3971-0 2_scaffold_2557-0 1_scaffold_6859-1 3_contig92075 2_scaffold_3040-0 2_scaffold_14221-0 3_scaffold_4633-1 2_scaffold_26226-0 2_scaffold_26254-0 3_scaffold_29161-0 ...the command line are as follows, you might noticed that we specified the -n and -p option, which means the number cutoff of transcripts assigned to a KO term and the corresponding FDR-adjust p value cutoff, respectively. the -s option specifies the prefix of the output result files.
$ python Hoea-version/src/Hoea/Hoea.py -i com5up.txt -a all.ko -s wahaha -n 0 -p 0.5also some information will print to the screen:
read in ho relationship file... done read in ho definition file... done read in background ho annotations... done generate ho term to gene id mapping,this may take a little while... done start analysis... input 336 items for analysis, 158 items with annotations 55195 items have annotations in background All Done!file generated are listed below:
wahaha_KO_1.dot wahaha_KO_2.pdf wahaha_KO_3.png wahaha_KO_5.dot wahaha_KO_6.pdf wahaha_Rinput.txt wahaha_KO_1.pdf wahaha_KO_2.png wahaha_KO_4.dot wahaha_KO_5.pdf wahaha_KO_6.png wahaha_Routput.txt wahaha_KO_1.png wahaha_KO_3.dot wahaha_KO_4.pdf wahaha_KO_5.png wahaha_Rcom.R wahaha_KO_2.dot wahaha_KO_3.pdf wahaha_KO_4.png wahaha_KO_6.dot wahaha_Rcom.Routas the first class of KO hierarchical structural contaons 6 terms, this will generate 6 graphs.
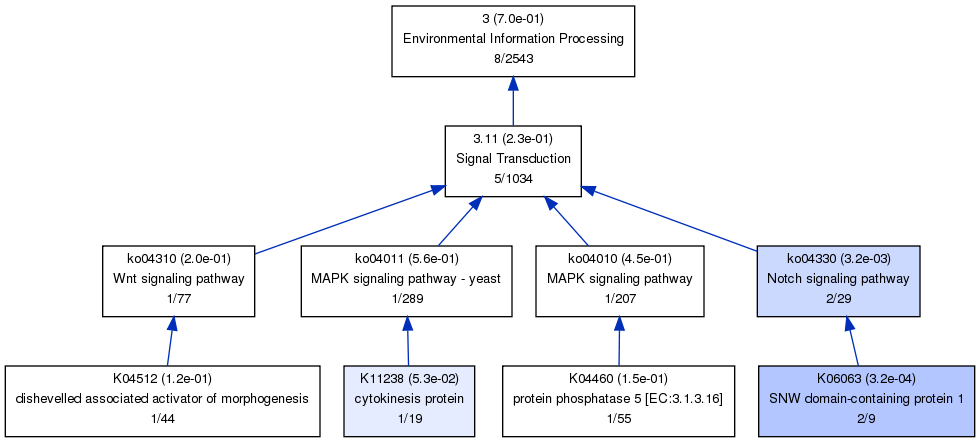
as the relationships between KO terms are not specified as GO like,so the terms were all connected with blue lines (which mean 'is_a' in GO).